Poster Abstracts
DEVELOPENT OF A COARSE-GRAINED POLYOXYETHYLENE GLYCOL NON-IONIC SURFACTANT MODEL USING THE SAFT-γ MIE FORCE-FIELD FOR MOLECULAR DYNAMICS SIMULATIONS - poster
Emma C L Richards, Erich A Müller and George Jackson
Department of Chemical Engineering, Imperial College London, London, UK
Robust coarse-grained (CG) models which exhibit both quantitative accuracy and representability are needed if one expects to obtain valuable insights to complex fluid systems. Within this study the SAFT-γ force-field methodology previously introduced in our group is employed to develop a CG surfactant model for use in the molecular simulation of non-ionic alkyl poly(oxyethylene) glycols (POE). In this approach, the effective Mie (generalized Lennard-Jones) interactions between the CG beads are estimated directly from target macroscopic thermodynamic properties with the aid of the molecular-based SAFT-γ Mie equation of state. Aqueous mixtures of alkyl poly(oxyethylene) glycol surfactants, CiEj , are considered as prototypical systems, where the interactions between the CG constitutive chemical moieties (alkyl, ether and hydroxyl groups), and their interactions with water are parameterised using appropriate experimental data for the vapour-liquid equilibria, liquid-liquid equilibria, enthalpy of mixing, and interfacial tension of selected pure components and mixtures. Here, a breakdown of the development and theory of the model will be discussed, as well as discussion of future applications such as the study of rheology and mesophase morphology.
SAFT γ-MIE COARSE-GRAINED FORCEFIELD FOR THE SIMULATION OF ANIONIC SURFACTANTS: PHASE BEHAVIOUR - poster
Matthias Kiesel, George Jackson, Erich A. Müller, Amparo Galindo
Department of Chemical Engineering, Centre for Process Systems Engineering, Institute for Molecular Science and Engineering, Imperial College London, London SW7 2AZ, U.K
In order to describe and predict the phase behaviour of amphiphilic molecules with standard molecular simulation, a balance must be made between an accurate description at the molecular level and computational cost. In previous works, forcefields based on the SAFT γ-Mie equation of state 1,2,3 have been derived. A major benefit of this top-down approach is the analytical parameterization, which facilitates the inclusion of a wide range of experimental data, delivering increased transferability and representability to the model. In this work, we extend our SAFT γ-Mie forcefield development workflow to develop models for charged species. Strong electrolytes are modelled as Mie segments with central point charges, interacting via a screened coulomb potential. Interactions between electrolytes and a water model2 are incorporated in a Mie potential, optimised to reproduce osmotic coefficients. The model can reproduce osmotic coefficients and densities of solvated electrolytes over the concentration and temperature range of interest.
1: Papaioannou et al., J. Chem. Phys. 140, 054107 (2014)
2: Lobanova et al., Mol. Phys., 113:9-10, 1228-1249 (2015)
3: Rahman et al., J. Phys. Chem. B 122, 9161−9177 (2018)
FORCE FIELD ASSESSMENT IN THE MOLECULAR SIMULATION OF INTERFACIAL TENSIONS OF SURFACTANTS - poster
Harry Cárdenas1, Siti Fatihah Salleh2, Sara Shahruddin2, and Erich A. Müller1
1Chemical Engineering Department, Imperial College London, UK.
2Specialty Chemicals Technology, PETRONAS Research Sdn Bhd, MALAYSIA
With advancement in algorithms and computational power, it is now possible to practically model systems that closely resemble those studied in the laboratory using atomistic molecular dynamics simulations. However, the impressive visual graphics and the detailed molecular insight obtained from the calculations distract from the fact that these representations and their quantitative predictions are only as good as the quality of the underlying force fields used to describe the intermolecular interactions. Hence, it is critical to identify the right intermolecular potential with the appropriate balance of accuracy, transferability and representability. We present here an example to demonstrate the impact of force field selection for surfactant performance assessment.
The systems under study herein include alkyl polyglucosides (APG) molecules; biocompatible and degradable surfactants formed by a sugar-based hydrophilic head, and a hydrocarbon-based hydrophobic tail. Two different interfacial environments are considered: water/air interface and water/oil interface, where the oil is phase is represented by n-decane molecules. Our initial simulations show that the pcff+ water model induces a physically unrealistic behaviour for the surfactants at the interface, particularly noticeable at the water-air interface (Figure 1) which are improved by employing an older model, (labelled here as pcff). The work discusses these issues in depth and served to highlight the general question of how much confidence we can/should have on the predictions of molecular dynamics simulations and the importance of testing the validity of the potentials against experimental results.
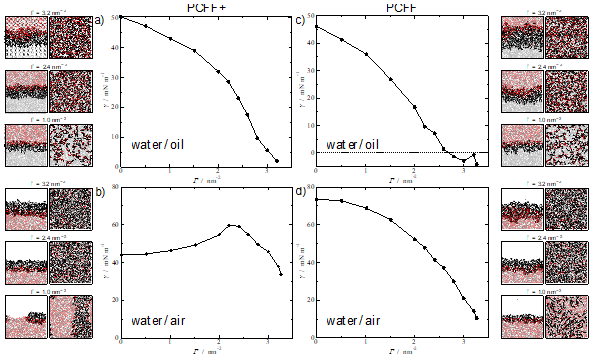
Figure 1. Interfacial tension at 298.15 K of: (top) APG12 at water/decane interface; (bottom) APG12 at water/air interface. The left figures correspond to the use of the pcff+ water model while those on the right with the older pcff water model. APG12 and n-decane molecules were modelled with pcff+ for all the systems. Snapshots for three different surface coverages values are added for each system, showing side and top view.
COMPUTER SIMULATIONS OF EPOXY BINDING ON IRON OXIDE SURFACES - poster
Charlie Wand1, Simon Gibbon2 and Flor Siperstein1
1 Department of Chemical Engineering and Analytical Science, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK
2 AkzoNobel Research & Development, Northallerton, North Yorkshire, DL7 7BJ, UK
Epoxy resins are widely used in protective coatings due to their good heat and chemical resistance, favourable mechanical properties and good adhesion to a range of substrates. As such, epoxy resins have been formulated as a protective coating for a wide range of applications, from aerospace and marine applications through to nontoxic interior coatings in the food industry [1]. In all cases, the performance of the final solid-polymer system is dependent on the physicochemical properties of the interface and the interaction between the polymer and the solid substrate. However, experimental methods to characterize this interaction are limited and mostly deteriorative to the interface. Computer modelling provides a tool to investigate the surface-polymer interface at an atomistic level.
Here we perform atomistic molecular dynamics simulations to investigate the binding of a common component in epoxy resins, diglycidyl ether of bisphenol A (DGEBA), on Iron Oxide surfaces (Figure 1) and investigate the effect of number of repeat units in DGEBA on the binding energy (ΔEbinding) defined as;
ΔEbinding= Eadsorbate/surface-(Eadsorbate+Esurface )
Where Eadsorbate/surface is the energy of the adsorbed DGEBA on the surface and Eadsorbate, Esurface are the energy of the adsorbate and surface in vacuum respectively. In epoxy resin applications the composition of the solid substrate is highly varied, with pre-treatments and production processes leading to a non-uniform surface chemistry and roughness. To reflect this, we investigate two Iron Oxides surfaces, hematite (Fe2O3) and magnetite (Fe3O4). We find that binding is stronger for DGEBA on hematite than magnetite, in agreement with previous literature findings [2] and suggest causes of this trend based on the surface termination.

This work was done with support from the EPSRC Prosperity Partnership SusCORD (EP/S004963/1).
[1] Friedrich, Jörg. Metal-Polymer Systems: Interface Design and Chemical Bonding. John Wiley & Sons, 2017.
[2] Bahlakeh, Ghasem, et al J. Phys. Chem. C 120 20 (2016): 11014-11026.
LEVERAGING FIT TO PURPOSE DRUG RELEASE MODELS TO HELP WITH FORMULATION DEVELOPMENT AND SCALE-UP EFFORTS - poster
Ravichandra Palaparthi, PhD[a,b]
[a]Anagha Consultants, Hyderabad 500050 India, [b]Anagha Consultants LLC, Hockessin DE 19707, United States
The quality of a product produced from a process is dependent on how it is formulated (the ingredients, their composition), but also on the process conditions of production, and the characteristics of the equipment used. Understanding of the interaction of these different factors, in addition to how the ingredients of the formulation interact with each other is important to achieve a consistent product quality. This is especially true for the pharma and specialty materials industry, where consistently maintaining finished product quality is critical. Hence formulators need to account for these interactions early on in their formulation development efforts and ensure they go hand in hand with the process/scale-up ones. A combination of fit for purpose process and application specific models can be leveraged to get actionable insights into the necessary experimentation to meet drug product quality attributes.
This poster and video focus on an example case study in this direction from the pharma space of sustained release microspheres products. It shows how a model for drug release [1] can be leveraged to bring insights into necessary experimentation to troubleshoot issues with variability in finished product's drug release rates. Use of such customized application specific models along with process models can help provide predictability to (and address troubleshoot such issues during) scale-up. Such customized tools facilitate the scientists and engineers in the industry to effectively leverage the power of simulations in their routine needs.
1. Mehan, et al AIChE Annual Conference, 2016